
In defense of RNASeq

We only use DNA and RNA for defining a cell (whether it’s a patient cell, PDX or cell line). I was asked many times about using proteomics data. In particular: why are we not using proteomics for modeling? Why do we think RNASeq is deep enough to get to a good model?
Information flow in a cell
Because - as the argument goes - RNASeq only tells you something about the rate of change in the proteome. It does not tell you a lot about protein concentration, let alone protein activity which eventually defines the phenotype - what the cell actually does.
Given that this is how information flows in a cell, the reasoning makes sense:

So - the logic follows - you could work your way backwards through the chain of causality and guess what the genome or the epigenome looks like based on how they impacted the transcriptome. Same way, you can guess the transcriptome based on the proteome.
But the other way is impossible. Since there is separate regulation at the proteome level, a single transcriptome can produce many different proteomes.
This is the gist of the argument. And I would agree - would these methods be at the same technology level. Sadly, (phospho)proteomics doesn’t really scale (for the time being). Proteins don’t readily amplify like nucleic acids do, and sequencers are much higher throughput than mass spectrometers.
So the question is: would you choose one proteomic sample over a thousand RNA samples (like a Perturb-Seq study)?
It depends. Is it even theoretically possible to figure out proteomics from RNA?
If not, then we need to go deeper, no matter the cost.
Defining cells with proteomics
First, it’s generally accepted that RNA levels correlate with protein levels in the steady state [1]. There is a good reason for that: proteins constantly break down and thus need to be constantly replenished via transcription, independent from anything else that may happen inside the cell. This results in a baseline correlation in around 0.6 (Pearson) between RNA and protein levels in the steady state.
So RNA could be good enough to initialize the model state.
Is it good enough?
The CCLE consortium has released a rather large-scale proteomic dataset[2] which we tested early 2023.
We trained three random forest models, one only using RNA data, one adding proteomics and one using both RNA and proteomics1.
Here they are:

The improvement is slight at best. Now we’re not huge experts in proteomics, so we may not have used the data the best way, but based on this data it certainly does not merit the 1000 to 1 cost of generating proteomics - for us, for the time being, of course.
Reverse engineering protein logic
All this was steady-state, initialization data.
But could we use transcriptomics to figure out the unknown logic of protein regulation?
Establishing causality requires running perturbation experiments. We are specifically interested reading out how the cell changes in response to perturbation.
In this case the steady-state assumption no longer holds, RNA and proteomics will be nowhere close to each other.
Still, we happily use tens of thousands of perturbation RNA signatures (differential expression vectors).
Why does this work?
Because the information flow is actually a loop.
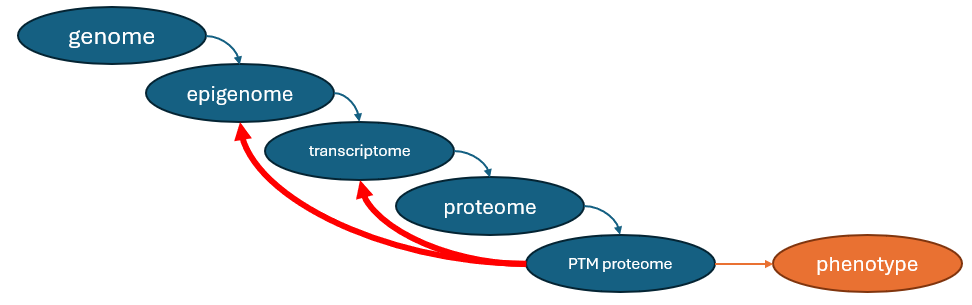
It works, because perturbations (ex. drugs) cause a cascade of protein activity changes, which - cascading down through the transcription factors - cause a host of transcriptomic changes.
It may be hard for us to deconvolute what pattern belongs to which change, but this is exactly what machine learning was invented for. Neural networks are great pattern matchers, even our own squishy ones. Can you tell me what animals these footprints belong to?

Easily, right? None of these footprints contain anything that actually makes an elephant or a human, but given the constraints that it must be an animal there is not much ambiguity.
These RNA footprints (a term coined by Schubert et al. [3]) gave us the opportunity to reverse engineer protein activity from RNA.
A case for multimodality
OK, how about having proteomics besides RNASeq? Surely it would increase the model performance, right?
Yes, it could. Take the following example:

Why is protein A inactive? Is it because it is inhibited (left scenario), or just because the gene is damaged (right scenario)?

These cases are theoretically impossible to distinguish unless given more data. Same goes mutatis mutandis for any potential pair of layers.
However, you could figure out the right gene regulation pattern still only using RNA if you add one more perturbed sample.

If knocking out gene B makes the protein turn on in the second study, we were observing the left scenario, otherwise we were observing the right one.
Of course, having another view at the system will also help noise correction, but, similarly, so could another sample. So this is my current opinion:
Multimodality helps, but only if it doesn’t cost much more than another round of samples.
This is why we use DNA as a second mode. DNA sequencing is also cheap, and while many DNA changes could be reverse engineered just by looking at the transcribed RNA, not all of them. As the example above shows, if a protein is not expressed at all, you would need additional experiments to decipher whether the underlying mechanism is specific downregulation of that protein or DNA damage.
Thank you Tamas Beke for the proteomics trainings!
References
[1] Liu et al. “On the Dependency of Cellular Protein Levels on mRNA Abundance”
[2] Nusinow et al. “Quantitative Proteomics of the Cancer Cell Line Encyclopedia”
[3] Broad “DepMap 2022 Q2 Public”
[4] Ghandi et al. “Next-generation characterization of the Cancer Cell Line Encyclopedia”
[5] Schubert et al. “Perturbation-response genes reveal signaling footprints in cancer gene expression”
RNA and gene sensitivity data comes from the DepMap 2022 Q2 release [3].