
The Patient Prediction Puzzle
on why solving patients need more than patients

OK, we need to talk about patient response predictions.
The baseline is surprisingly high
There’s a striking disconnect between how pharma insiders view AI’s relevance in clinical trials (close to zero) and how it’s often marketed (AI is already here! 70% accuracy!).
If someone tells you they can predict how patients will respond to a drug with 70% accuracy, how surprised should you be? Given that most phase 2 trials - where efficacy is first measured in humans - fail, that sounds like a big leap forward, right?
Let’s test that.
The gold standard for fully mapped patient data is the Cancer Genome Atlas (TCGA). The team found around 800 data points where we know all three of:
the patient’s molecular profile before the treatment
the treatment itself
the outcome
On first approach, how does a simple linear regression do?
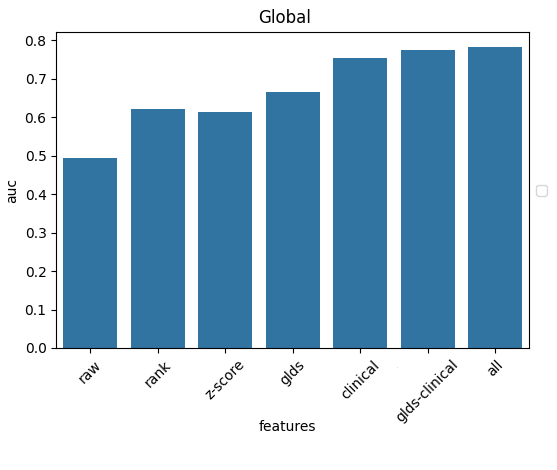
Wait, 75% already?
What’s the real question?
By predicting clinical patient response data, the question you’re really asking is:
Can you predict how an approved drug will work on a patient treated according to protocol?
But this data contains very little molecular information. The best-performing indications and biomarkers are already built into the treatment protocol, and doctors rarely go off-script.
No wonder the only meaningful variables that show up are the cancer’s indication, its stage, and the drug used - essentially, the survival statistics of the state-of-the-art.
That’s not the question we actually want to answer.
The real question for getting a new drug into the clinic is:
Can you predict how well a new, never-before-tested drug will work in different indications and where could it beat today’s standard of care1?
That question, almost by definition, can’t be answered from existing patient data. You only get 2-3 data points per patient, and from a very limited set of drugs. There’s simply no way to infer what would have worked.
More data?
Maybe the problem is just scale. Let’s add more data. Why just 800 data points, anyway? There are hundreds of thousands of patient data points out there.
On one hand, good data is surprisingly scarce.
We started with 12,000 samples from TCGA and 200,000 from GENIE, but it quickly started eroding.
Most samples had no response data at all.
Many represented early-stage cancers removed surgically - no way to tell whether the drug was even necessary.
Others were sequenced after treatment, so the tumor’s molecular profile represents the recurrent tumor, not the one you treated.
After cleaning, we were left with about 800 usable data points from TCGA and 3,000 from GENIE.
In theory, with unlimited data, the problem becomes solvable: for every patient who doesn’t respond to a given drug, there would eventually be others similar enough who responded to a different drug. At that point, an AI model could start connecting the genetic dots.
That’s exactly the hypothesis we’re testing now in collaboration with Memorial Sloan Kettering’s Cancer Center in this year’s iHub Challenge program - how much additional signal would emerge if we had an order of magnitude more data?
Huge thanks to Rick Peng, Jae Zhong and the MSK team for making this possible.
Building bridges
Still, my intuition is that patient data will remain just one piece of the puzzle. To reach the right level of molecular understanding, we’ll likely need to mix in data with much higher information density - where we can actually generate true counterfactuals (what-ifs).
That means simpler, but repeatable models like in vitro, organoid or PDX (patient-derived xenograft) models.
Explainer box: The acronym PDX stands for Patient-Derived Xenograft. By implanting human tumor cells into mice, we can test drugs not yet approved for clinical trials on fresh human cells, in a tumor-like living environment without compromising patient safety (unfortunately, the same can't be said for the mice)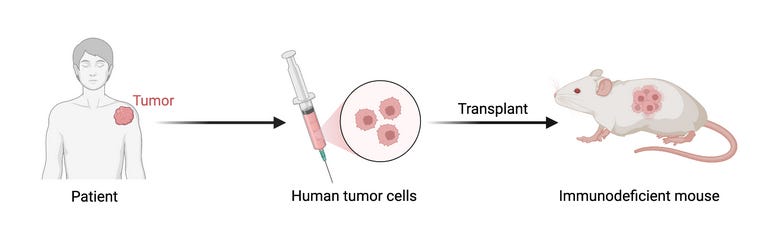
But how do we bridge these worlds?
An in vitro cell line doesn’t map directly to a patient. If you change both the treatment and the input system simultaneously, you can’t tell whether the outcome came from the drug or the model.
Turns out there might be a bridge. It’s faint and thin for now, but it could just become strong enough if we put the work in. See the following plot:
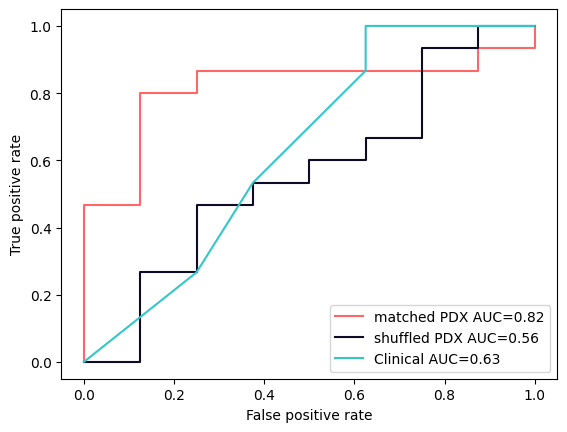
PDX models show a predictive signal above that of the clinical baseline - but only when matched to their original patient. Just the same indication isn’t enough.
Now all our studies are very preliminary, hundreds of patients at best. The image above is made from just 23 patients.
That’s because there isn’t much data that meets these strict criteria, especially publicly. That’s why I’m glad to have our collaboration with Champions Oncology (shout-out to Matt Newman for helping us make this happen!). Together, we’re expanding that bridge to thousands of data points.
If it holds, this connection could let us integrate all the “unmatched” PDX datasets as well into one coherent predictive system.
I’ll be back with details when we get there.
Kudos to the many people on the Turbine team who made the studies and these collaborations possible: Richard Izrael for leading the Champions partnership, Emese Sallai-Simon for supporting us during the MSK process, Gema Sanz, Bence Czako, Richard Izrael and Bence Szalai for the PDX/patient studies!
while not being unreasonably toxic, of course