
Make multimodality work for you, not against
What are we assuming in a multi-modal training? What can you measure?

Large-scale RNA data generation is starting to show diminishing returns. So, naturally, multimodality is starting to look very attractive.
After all, RNA is just one slice of reality. If we want to be able to build a general virtual cell, RNA alone may not be enough. So it makes sense to collect and add other types of data out there. More data could only improve our results, right?

Well, not necessarily. If you just mindlessly add multiple types of data to a training, it can just blow up, drastically decreasing performance.
A case in point
One of our teams was recently training a new model to power virtual assays for drug target discovery.
We didn’t just want to predict the overall effect of knocking out a gene, DepMap already measured that. We wanted something that can predict what happens inside the cell when that gene is knocked out.
A DepMap-wide Perturb-seq, if you will.
The obvious starting point for the training was to add DepMap and Perturb-Seq data together, and see what happens.
This.
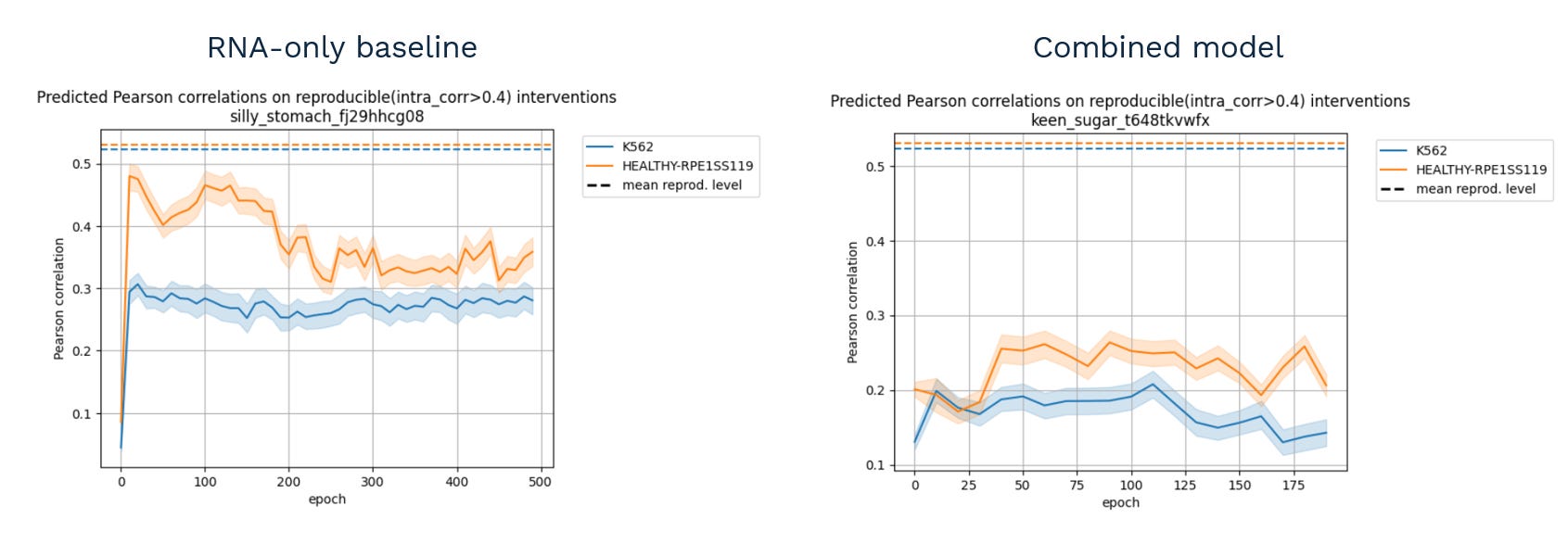
That is, instead of improving (or, in the worst case, doing nothing), the performance dropped significantly when the model tried to combine the two data types.
After spending some time chasing the culprit (as the two datasets are hard to compare due to being different readouts), the team came up with the following plot:
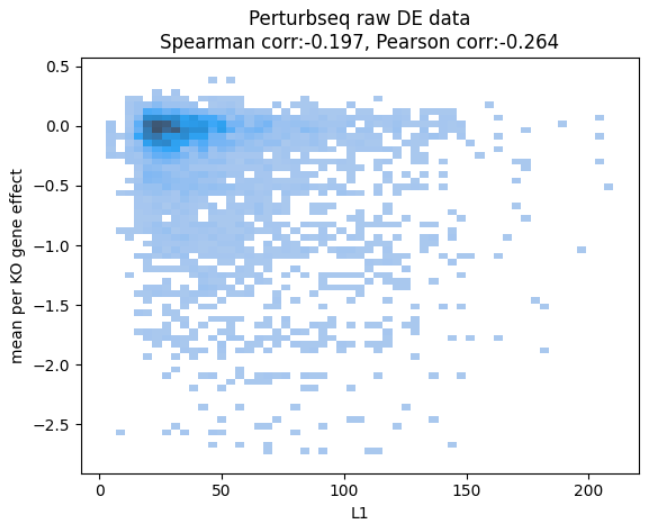
revealing that, even in the ground truth data, how much the RNA levels change and how the cells fare seem quite unrelated.
This doesn’t make too much sense. Surely knocking out a gene that eventually kills the cell should start a cascade of repair processes which should be visible in RNA.
Either our metric (using L1 norm) is wrong, or the data doesn’t line up - that is, the biology is very different.
Different biology was a plausible explanation as the gene effects are measured in a 21-day assay, while the Perturb-seq experiments were generated in different labs, different setup, 7 days after transduction.
But if there’s no shared biology at all, there’s no way to connect these modalities together.
We got lucky. In the end, what was missing is a simple correction on cell counts:
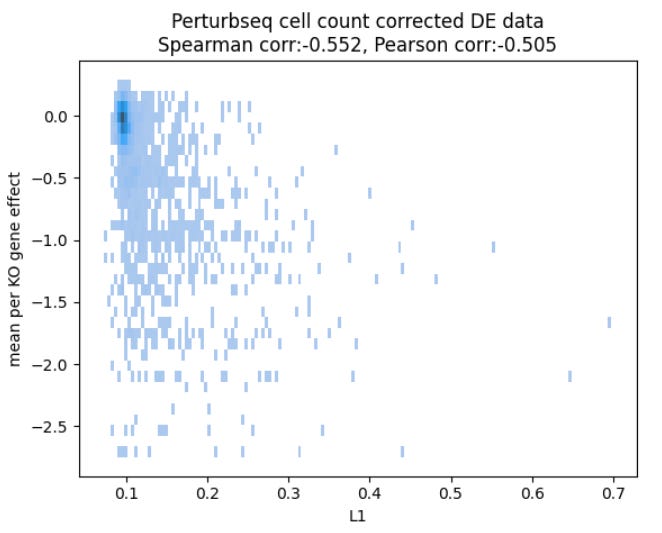
and after training on the corrected data, we got all the missing performance back, maybe1 even some more:
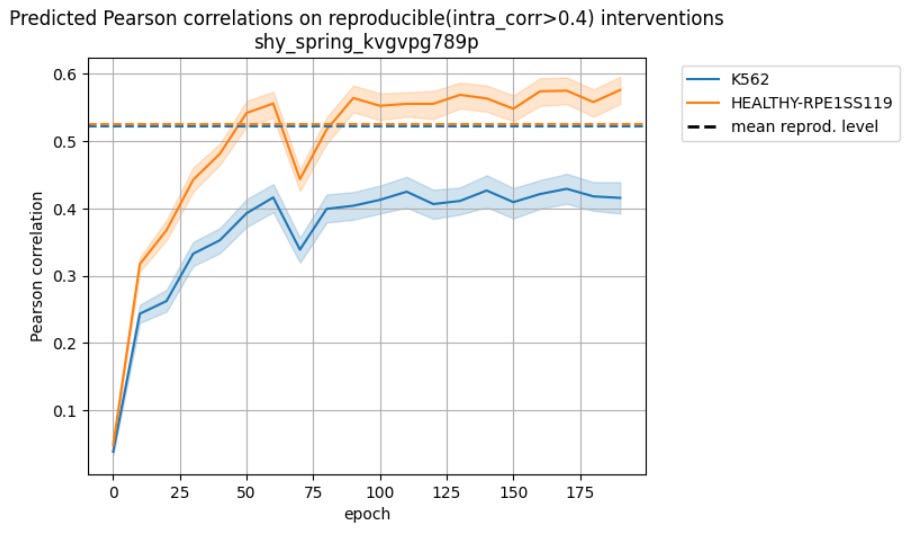
So there was a way to make the modalities work together despite the obvious differences in protocol.
That’s what seems to be the core question at play when building something multimodal: am I measuring the same biology when putting two datasets together?
The elephant in the room
Which got me thinking. Is there a way to generally quantify how good your assumption of shared biology is?
Imagine biology as cells moving on some kind of latent manifold describing the internal cell state. Then every modality is a projection, a lower dimensional representation of that mathematical surface.
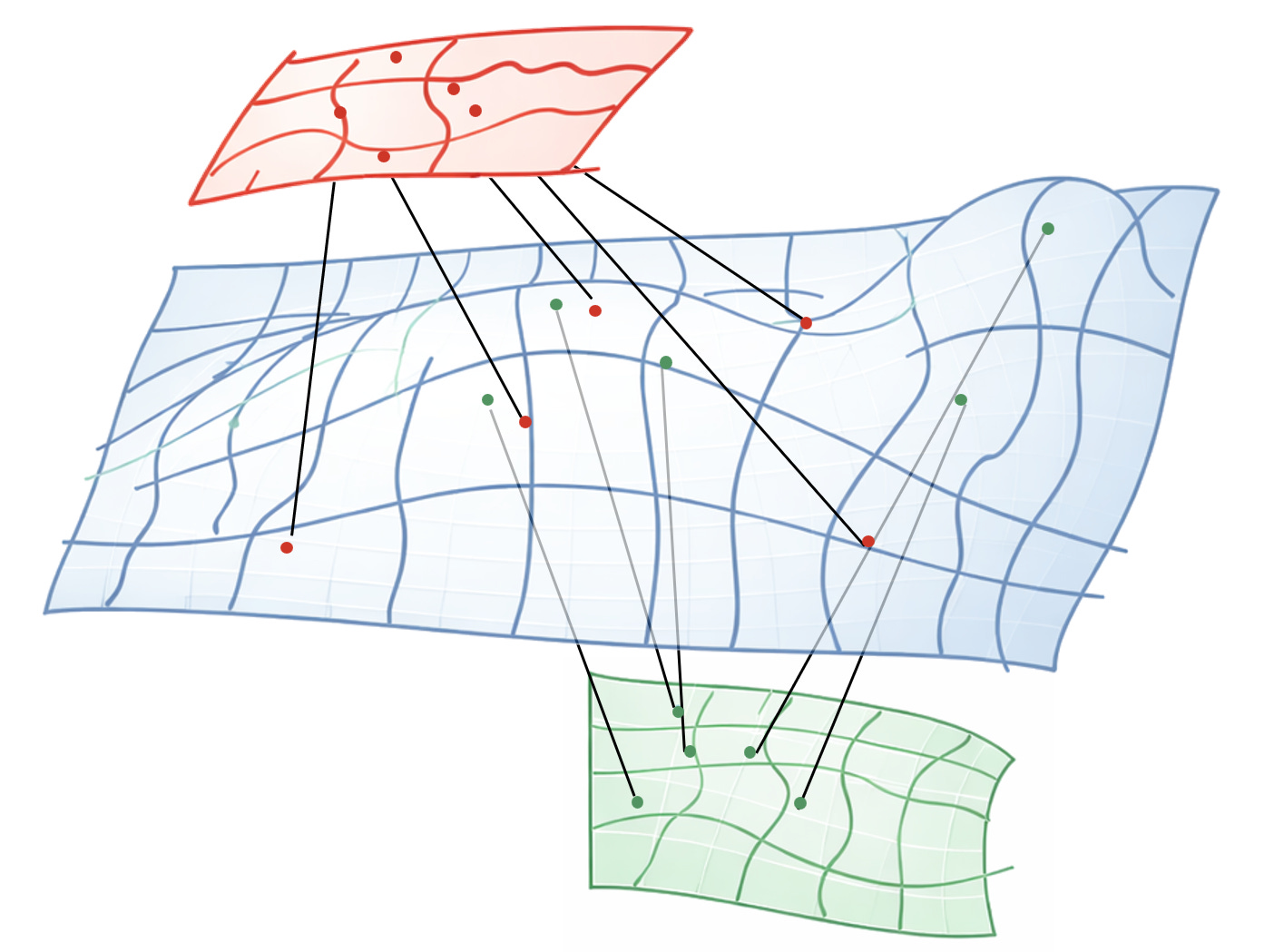
You can collect as many projections as your budget allows, but you never get to see the surface itself. Hence the elephant and the blind men.
In theory, if your surface is smooth, and your sampling is dense enough, you could reconstruct the blue surface close enough to recover the true “working manifold” of biology.
You know you’re close when you can reliably predict across all modalities from just a few of them - RNA to phenotype, proteome to epigenome, etc.
But to do that, you need to learn which regions of the green space are close to which regions of the red space in the original blue space.
Which means points that are somehow externally paired in the training data.
That’s why Ron argued so passionately about paired data.
Truly, in an ideal world, every sample would come with every modality.
But most molecular measurements are destructive. So every pairing decision is fundamentally an assumption of shared biology — an assumption that the two measurements originate from the same spot in the blue surface.
It’s a stronger assumption if the data comes from the same patient, lab and protocol, but an assumption nonetheless.
On the other hand, sometimes data that look wildly unpaired can still work, as the example above shows.
So what actually makes multi-modal data work? I think it’s the following assumptions.
If the data doesn’t exist, following these could be a structured way to build it, testing each assumption as you iterate on your protocols.
If the data already exist, maybe it’s worth trying the opposite: train first. If it fails, start tracing back the chain of assumptions and see whether there’s something you can fix.
The pairing assumption
The deepest assumption is that each multi-modal pair truly describes the same biological state. The test is this: can you produce matching pairs with a single readout?
You are pairing samples based on them being in the same experimental condition? Or you are matching individual cells in heterogeneous assays based on microscopic features?
Both could be valid pairing algorithms. But in your setup, do they really produce consistent RNA-Seq vectors, before considering any other modality?
If they are consistent to your liking, it’s now safer to assume the other readouts from the same batch will be consistent as well (come from the same distribution).
If not, your pairing is off. Maybe you are missing experimental variables to control (synchronizing the cells, for example). Maybe you need better markers to match cells in imaging.
The smoothness assumption
Next up the ladder is checking if you have enough data to approximate the biological surface. The test: Can you reliably predict one modality from the others? Under which conditions?
Suppose you have some RNA-Seq data points and immunhistochemistry (IHC) images. Let’s say they come from different patients, but some are matched by indication, so maybe they are close enough.
If you can reliably predict one modality from another, it means your sampling density is sufficient relative to the smoothness of the surface between those points.
Just don’t forget to test how far that generalizes - Crossing tissue types is the barrier where it usually breaks.
If the prediction doesn’t work at all, or the training blows up, then the data points are too few and far apart (or, if you’re coming from the opposite direction, they may not represent the same biology).
Extrapolating
If all you wanted was something that translates from modality A to modality B, you’re done once the above prediction works. Usually we are asked to predict responses for new drugs or cell lines. Those responses won’t have any of the modalities measured.
In the end, the arbiter is whether your model improved by being multi-modal. Did you gain the ability to extrapolate to new interventions?
Because that’s why we ultimately generate all this data - to learn enough about the hidden blue surface to trace the missing parts.
Because that’s where the cures for disease lie.
Thanks to Laszlo Mero, Milan Sztilkovics and Zsolt Gyure for the case study data, and Daniel Veres for the review and comments!
There is a clear improvement, but ablation studies show that most of it is driven by the correction, not the integration.