
What can Virtual Cells do for you today?
and what they can't - because the bottleneck is not computing power

The term Virtual Cell has taken off over the past year. On one hand, it’s great to see so much attention on computational biology. On the other, it has created confusion about what current and emerging technologies can actually do.
Let me try to clear that up.
What is a virtual cell?
The current trend around virtual cells traces back to this article by Bunne et al., where they introduced the term AIVC, or AI Virtual Cell. According to their definition:
In particular, an AIVC needs to have capabilities that allows researchers to (1) create a universal representation (UR) of biological states across species, modalities, datasets, and contexts, including cell types, developmental stages, and external conditions; (2) predict cellular function, behavior, and dynamics, as well as uncover the underlying mechanisms; and (3) perform in silico experiments to generate and test new scientific hypotheses and guide data collection to efficiently expand the virtual cell’s abilities.
In other words, a virtual cell would be a complete, general simulation - one that can accurately predict biology in new contexts without retraining.
That’s the dream.
Explainer box on biological context
Biological context is an umbrella term for all of the known or unknown variables and interactions that influence an experiment's outcome but are not explicitly measured.
The same cell can respond differently depending on:
- neighboring cells
- extracellular environment
- experimental protocol
- the natural evolution of the cell line over time
- or anything like the current moon phase (I kid you not)
which then become implicit conditions on any biological measurement. (Drug A is effective in X cells, but only if... (after X days, without support cells close enough, etc.)The Bunne et al. paper states that the main bottleneck to achieving this general virtual cell is integration of diverse data types and scales. Some readers interpreted this as suggesting that, with the right (foundation) modeling approach, we could build a general virtual cell from the data we already have,
which is where my opinion differs.
Explainer box on foundation models vs virtual cells
It’s easy to confuse these terms because they’re often mentioned together, but they are different concepts.
Foundation modeling is a method. AI models trained first on a massive, messy, unlabeled (in our case: has no specific treatment or outcome target, they are just there) dataset to understand the general problem space. Then, with a smaller set of task-specific data, they can be fine-tuned to perform specific tasks. This is how today's (Transformer-based) large language models work.
A virtual cell is an application. Virtual cells are models that predict how a biological system will respond to novel inputs. Virtual cells can use any type of model internally from a simple linear regression to a deep neural network like a Transformer.
So you can have virtual cells that aren't foundation models and foundation models that aren't virtual cells.
But foundation models are probably our current best bet for moving toward that elusive general virtual cell.Understanding context will enable general virtual cells
If we want models that generalize to new biological contexts, we first need to understand what context actually means. You can’t predict outcomes in a new situation without knowing whether your training data can tell you that the situation is different.
We don’t need to list every single variable that influences an experiment - there are too many - but we do need cell “snapshots” rich enough to capture the fingerprints of all meaningful influences.
Put simply: if the same input can lead to multiple outcomes (beyond random noise), then the input is incomplete.
In theory, a perfect RNA-seq dataset might capture all this information, since most biological processes leave some trace in RNA levels. In practice, though, today’s signal-to-noise ratio likely erases many of those traces.
This probably isn’t a fundamental limit of biology, but of our experimental tools and how we use the data. Because
biology itself is remarkably reproducible; monozygotic twins (and clones) prove that you can replay embryonic development with great fidelity.
Yet, we struggle even trying to combine data from different labs or protocols.
So to build a general virtual cell, the first thing - I think - we’d need is a numeric description of cells detailed enough that no additional biological context needs to be specified. A digital cell, if you’d like.
This isn’t entirely out of reach, but I don’t think any data we currently have (in the forms we usually process them) carries that level of context.
So for general virtual cells, rather than throwing more compute at existing datasets, the better path forward is to determine which kinds of data actually enable predictions across new biological contexts, and start generating that.
The virtual cells we already have
Today’s data and methods give us is narrow virtual cells. Having calibration data for a well-defined assay, we can already virtualize many laboratory experiments with impressive accuracy. We call these virtual assays.
When we looked at which experiments could be simulated most reliably, one pattern stood out: success didn’t depend on the type of perturbation (drug, CRISPR, RNAi) or on the specific endpoint (viability, transcriptomics, phenotype). It depended almost entirely on the nature of existing data from that assay. Like this:

A few interesting observations from this.
Virtual cells can already help scale your experiments. You can run hundreds of millions of simulated experiments, choose the best ones, and the majority of them will work as expected1 (if you have previous training data for your drug in which case you are in one of the bottom rows).
Adding new perturbations without training data makes the problem much harder. There is real signal in the simulations, but you need specific expertise to untangle it from the noise. This is the region where a good model and clean data can make the biggest difference.
You can only be as good as the underlying assay is. Some wet-lab assays are bad at reproducing their own results. I’m looking at you, Bliss synergy2. I imagine as we grow in our understanding of what are the constituents of biological context, we can design experiments that replicate better.
Most foundation model tests target the easy problems. The currently running Virtual Cell Challenge provides training data that already includes the exact cell line and perturbations being tested. There is not much benefit to be gained here by heavy-duty foundation models compared to simple ML models.
The “AGI tests” of biology
So, we are, for the time being, in the narrow era. Every new advance is appreciated, but be careful - it’s easy to claim far too wide and general usability for the next narrow win.
I think there are some biological problems which can’t be solved without genuine context transfer. Mastering those would mean our models have begun to understand biology itself.
If you wanted to define an “AGI3 test” for biology, the ladder of progress could look like this:
Take a new drug with known structure and binding data and predict how cells will respond using only their CRISPR response.
Predict drug synergy between two compounds that have only been tested in monotherapy before.
Predict how two cell types, previously tested alone, respond when co-cultured.
Predict how a drug tested only in vitro will behave in ex vivo or in vivo models.
These sound simple, but none - according to my knowledge - have been solved to experimental reproducibility4, not even close. Some models and approaches are useful, but far from reproducibility.
Still, step by step, as we expand the context distance and the time horizon of what our models can predict, we’ll eventually get to the ultimate test of biological understanding:
Given only the a DNA of a zygote, replay embryonic development until the baby is born.
The work of Laszlo Mero, Gerold Csendes, Murat Cem Kose, Dana Zemel and Robert Sipos were instrumental to get the data package together for the article. Also thanks for the reviews, Louisa Roberts, Krishna Bulusu, Daniel Veres, Imre Gaspar and Valer Kaszas!
I mean half of the signal will still be there. Interesting signals are rare. If you have 50% (coin-flip) accuracy, and 5% interesting signal, most likely all of your top 10 experiments will fail. You need more than 90% accuracy on the average data point to keep half your signal in the wet-lab. Here’s an illustrative example below (with 6% signal to keep numbers nice).
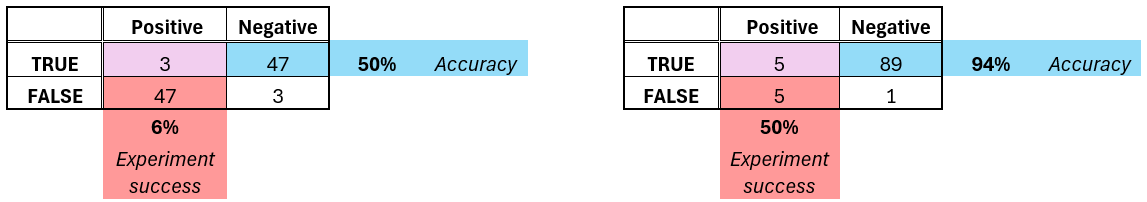
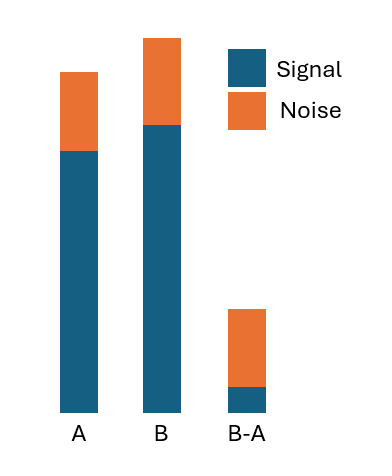
Combination synergy metrics, like Bliss, are famous for being noisy. It is because they are fundamentally difference metrics of the effectiveness of the drugs administered individually vs in combinations. A difference - especially if the two arguments are close - amplifies noise, sometimes extremely so. The figure below should give an intuition why that happens.
Artificial General Intelligence - simply put, the quest in general AI to get to a model which is really intelligent, not just sounds intelligent. I think it’s a good analogue, but otherwise it’s a huge can of worms I really don’t want to open here.
With a robust metric - I know I’m belaboring this point, but none of these are apparent if you’re using a simple metric. It will just give you a nice 70-90% accuracy, and you’ll be happy until the wet-lab experiments start failing. Since the outliers will be selected for lab validation, your model needs to work there, not just on the average data point.
Thanks, Andy, I'll try to make it easier to understand, good to have your feedback!
So that's the interesting part - by and large, just adding totally unrelated treatments doesn't really move the needle. But if there are some similarities - eg. having a similar mechanism, targeting the same protein or the same cellular pathway - there can be a good predictivity gain. This is a good example of how, in many cases, you can get ahead with the right expertise. It's just that at this stage, you still need to add brains to make it work.
Reading your articles is a difficult thing (not having a background), but the topics you chose are phenomenal. :) Thank you for putting in all the effort to crystalize and share your thoughts and make them available to anyone!
I know this may be a maybe not even valid question, but...
Just wondering, what would be the rating of a training setup for a virtual assay with exact cell, multiple different treatments? E.g. If I know how a cell reacted in the past to various other treatment scenarios, would I have a better chance of predicting the assay outcome for a new one? (Last Marginal in the table, but not with multiple treatments)